
Updates from ASH: How New Discoveries Will Impact Personalized Care in MPNs
Treatment News from the American Society of Hematology on Myeloproliferative Neoplasms
Hear about cutting-edge research and new therapies presented at the 2024 American Society of Hematology meeting. Learn how individualized treatments can improve your outcomes and quality of life. Get practical strategies for handling common side effects of MPN treatments. Discover innovative therapies, including JAK inhibitors and combination treatments. Find out how to stay informed and participate in promising clinical trials. Learn the key questions to ask your healthcare team to ensure you’re receiving the best, most current care.
Whether you’re newly diagnosed or managing ongoing care, learn how the latest findings impact your myeloproliferative neoplasms (MPN) treatment options and quality of life.
Dr. John Mascarenhas of The Tisch Cancer Institute at Mount Sinai and patient advocate Andrew Schorr share the latest breakthroughs in MPN care. Explore personalized treatments, cutting-edge therapies, and groundbreaking research that are changing how MPNs are treated. Learn how new discoveries can improve your treatment options, help manage side effects, and enhance your quality of life.

Thank you to The Leukemia & Lymphoma Society for their partnership. The Leukemia & Lymphoma Society is here for you with information about clinical trials, resources, and support.
This interview has been edited for clarity and length. This is not medical advice. Please consult with your healthcare provider to make treatment decisions.
- Introduction
- Is There Encouraging Progress for Myelofibrosis Patients?
- What Have We Learned from MPN Gene Mutations?
- How Do Doctors Choose the Right JAK Inhibitor for the Patient?
- Is Combination Therapy the Next Step in Building on JAK Inhibitors?
- Transplant vs. Medication: Where Do We Stand?
- Will PV or ET Progress to Myelofibrosis? What Can Be Done?
- Progression to Acute Myeloid Leukemia (AML)
- Are We Making Progress with Secondary AML in MPN Patients?
- Role of Interferon for the Treatment of MPNs
- New Drugs Being Studied in MPN Treatments Clinical Trials
- Considering a Clinical Trial
- Getting the Test Drug vs. a Placebo
- What If I Have Other Chronic Conditions?
- The Future of Treatments for MPN Patients
- What Patients Can Do to Alleviate Symptom Burden
- Final Takeaways on MPN Treatments
- Conclusion
Introduction
Tiffany Drummond: As a clinical researcher and patient advocate, I am excited to talk about some very exciting developments in MPN treatment, including important breakthroughs and promising new combination therapies. Many of these advancements were highlighted at the 2024 American Society of Hematology meeting, better known as ASH, where leading doctors and researchers from around the world gather to share the latest findings.
Our goal is to provide patients and care partners with valuable information to help in their healthcare journey. We want to empower you to have informed conversations with your medical team so you can better understand your treatment options and how to balance effective care with maintaining your quality of life.

We want to thank all of our promotional partners for their support. It is because of you our programs reach the audience who needs it. While we hope you find this program helpful, please keep in mind that the information provided is not a substitute for medical advice.
Let’s kick off another engaging conversation with amazing patient advocate Andrew Schorr and leading hematologist-oncologist Dr. John Mascarenhas.

Andrew Schorr: Welcome to this program about the latest in MPNs. I’m with a friend and leading scientist-physician Dr. John Mascarenhas at The Tisch Cancer Institute at Mount Sinai in New York. John, you have many titles. You’re a noted hematologist and subspecialist in MPNs. Thanks for joining us.
Dr. John Mascarenhas: Andy, thanks for inviting me. I always enjoy connecting with you.
There has been a lot of interest as we understand the disease biology even greater than we did in 2005 when the JAK mutation was first discovered.
Dr. John Mascarenhas
Is There Encouraging Progress for Myelofibrosis Patients?
Andrew: It’s very personal for me. I’ve been living with primary myelofibrosis since 2011 and it’s somewhat progressed. I’ve been on two JAK inhibitors and maybe I’ll switch to a third. Will I have combination therapy with a JAK inhibitor and something else? We all wonder about that.
Some of us are concerned. Should we have a transplant or can medical therapies take the place of a transplant? If you have polycythemia vera or essential thrombocythemia, you ask if you’re going to progress to myelofibrosis and at what rate. How is our situation different from the next person?
John, you were a speaker at the 2024 ASH meeting in San Diego, and you were involved in lots of studies. I want to talk about what’s significant for patients. We have the current therapies and a lot of drugs that many of us have never heard of that are in development. You’re involved in a lot of the development. Which way is the wind blowing? Are you encouraged for us? We saw progress in other blood cancers. Is it now starting to blossom in MPNs, specifically for myelofibrosis?

Dr. Mascarenhas: The short answer is yes, I am encouraged. That’s a fundamental defect that I have, continuing to be optimistic no matter what we’re looking at. That optimism has been maintained over almost 20 years that we continue to move in the right direction, but unfortunately, often not fast enough for our patients.
What I’ve seen is the evolution of the JAK inhibitor era, which you alluded to. We have four JAK inhibitors that are approved that allow us to tailor and personalize the therapy to patients based on their profile and blood counts, and even provide serial sequencing of JAK inhibitors. But that isn’t enough.

There has been a lot of interest as we understand the disease biology even greater than we did in 2005 when the JAK mutation was first discovered. We now recognize that there’s a greater degree of complexity and heterogeneity among patients. There are a lot of different pathways that appear to be very important and relevant to the physiology and pathophysiology of this disease.

There is a real interest in targeting the grandfather/grandmother cell in which the disease originated. We are looking for vulnerabilities in those pathways in that cell population that would allow us to ultimately delete that cell to provide deeper responses and even curative approaches. Outside of transplantation, the therapies we have don’t cure patients. They address issues that are not unimportant, like spleen size and cytopenia or low blood counts, and improve how patients do and ultimately prolong survival. But we’re looking to leverage these findings from the lab to find therapies that change the disease course and improve outcomes like survival and progression-free survival.

Many agents are leading us in that direction. These agents turn on the p53 pathway, like navtemadlin, an oral drug that binds a protein called MDM2 and relieves repression on p53, allowing for the natural cell processes to be induced, which is cell death of the malignant cell. It’s a fascinating concept. Navtemadlin is at the forefront of doing that. We showed data in the relapsed/refractory setting of using that as a single agent. We’re now going to move it up to combinations.

Drugs like that are telling. They’re hitting pathways that can induce malignant cells at their core to die, to synergize, and be practical with it. We want to create therapies and approaches that capitalize off the benefits we have, like JAK inhibitors, which can be well-tolerated but can provide deeper responses than what we’ve seen thus far. Navtemadlin is a great example of that.

What Have We Learned from MPN Gene Mutations?
Andrew: We’re going to go through a laundry list as we dig into different drugs, but I want to go over what you said. You’re trying to go back to the very basics of cancer, what went haywire in a patient who ends up with a bone marrow problem that leads to one of the MPNs. Can you shut it down at the earliest stage by understanding it?
Over the last few years, your scientific community has identified different oncogenes (cancer genes) that have been responsible for that. You talked about the heterogeneity or the differences. Some of us have CALR, some have JAK2, and some have MPL. Is that understanding helping?

Dr. Mascarenhas: I do think it helps because we recognize that it’s not a monolithic disease. The driver mutations and the different amounts of those mutations that are understood to be present in the bone marrow cells as well as the sequence in which the mutations arose all tell a picture. They paint a picture of complexity at the molecular and cellular level that explains why there’s heterogeneity at the clinical level — why some patients have very high white counts and some patients have very low platelet counts; why some patients have very big spleens and some patients have a lot more anemia and transfusion dependence. It can all be explained relative to the biology, these mutations, and the effects of these mutations.

Once we’ve realized that, the next step is how to take all of that complex data and distill that to help us understand how best to target those cells based on that genomic complexity. That’s where things like artificial intelligence and machine learning will help us move the field forward as it’s doing with other sciences. We’re moving in this direction of a deeper understanding of the biology from the molecular standpoint that informs us with prognostication, which is important, but most importantly, therapeutic implications.

There is substantial data that would suggest that certain mutations likely influence outcomes and responses to treatment. Most patients will have had next-generation sequencing. You look at these gene panels and see if mutations exist in any given individual and what they mean. We know that some of these mutations can have influenced prognosis and outcome. Some of these mutations and the presence of more than one mutation could even predict a lesser response to drugs like ruxolitinib, a less robust spleen, a shorter duration of response, and a quicker time to failure of drugs. Knowing that upfront may be strategic in understanding how better to approach diseases rather than give the same drug to everybody.

The same drug for everyone is not going to be the right answer. At the forefront, drugs that target CALR, for example, are exciting. We’re taking out a subset of patients with myelofibrosis and ET with the mutant CALR protein expressed on the surface of the cell and saying these patients may be best served by a drug that specifically targets that protein on the surface. That wouldn’t make sense for a JAK2-mutated patient. A JAK2-mutated patient may be best served with a small molecule inhibitor that specifically and selectively targets the JAK2 mutation, which is under investigation. You’re seeing these mutations inform the clinical development of very selective and specific drugs.

Andrew: I almost think of next-generation sequencing like a modern art painting with red and blue splattered. Hopefully, with some consultation with an MPN specialist, you can find out how current therapies or investigational ones apply to your specific situation.
The genes that are driving our illness may evolve, so what the story is today may not be the same story in a year, two, or three.
Andrew Schorr
How Do Doctors Choose the Right JAK Inhibitor for the Patient?
Andrew: You also mentioned how the genes that are driving our illness may evolve, so what the story is today may not be the same story in a year, two, or three. I’ve been living with myelofibrosis since 2011. It has been pretty stable and has been driven by JAK2 V617F. You mentioned the four current approved JAK inhibitors. They have nuances and it would seem like the choice of which one or sequencing, as you said, may vary by patient based on their situation. How do you know where to start?

Dr. Mascarenhas: We are blessed that we have choices today because it wasn’t always the case. We have opportunities to select drugs that may be best suited for different subsets of patients. For example, patients with low platelets or those who have cytopenic profiles may be best served by drugs that are easily delivered and have rationale in that patient population, namely pacritinib over ruxolitinib, in which we know platelets often limit the ability to dose up on ruxolitinib. Fedratinib, even more recently, had some data providing some more security there. We know that platelets could be a determinant of which one of the JAK inhibitors you’re going to select.
Most patients will start with ruxolitinib. It’s the oldest, most familiar, and probably still one of the best drugs that I’ve ever seen in this field
Dr. John Mascarenhas
Anemia is another one that’s gotten a lot of attention. It can be a major issue for patients at presentation and can worsen over time. Ruxolitinib is a great drug, but it can worsen anemia for some patients, so it may not be the best therapy in that setting. However, drugs like momelotinib or even pacritinib that inhibit ACVR1, a different protein, can improve anemia in some patients.

We use patient profiles to help us understand which drugs to choose from, but for the most part, most patients will start with ruxolitinib. It’s the oldest, most familiar, and probably still one of the best drugs that I’ve ever seen in this field in terms of achieving its goals. But again, not every patient fits the bill, so you can tweak that as needed.
Is Combination Therapy the Next Step in Building on JAK Inhibitors?
Andrew: A lot of studies talk about building on ruxolitinib and I’m sure there’s discussion about building on the other JAK inhibitors as well. Is a one-two punch necessarily better? Is that where you’re headed?
Dr. Mascarenhas: It’s definitely what we’re interested in asking. We have JAK inhibitors that are disposable and beneficial, but they’re not sufficient. We have other drugs that have demonstrated clinical activity and are rational and validated in preclinical models, which are systems that help us understand whether it makes sense to take it into a patient. These are drugs like pelabresib, imetelstat, navtemadlin, and selinexor, but each drug has a rationale and is active even as a single agent in these diseases.

The question being asked now is: is it better to combine the two drugs? If you look throughout oncology, most of oncology is treated with combinations of therapy. It’s very rare to find an oncologic disease, whether it’s of the blood or of the solid malignancy, where we use one agent and then if it fails, we go to a single subsequent agent. Usually, combinations of therapy are more potent together.
If you put agent X — whether it’s selinexor, navtemadlin, pelabresib, or imetelstat — together with ruxolitinib, which tends to be the first drug you pick, you tend to see better efficacy and, in some cases, even better safety profile when the two are combined.
Dr. John Mascarenhas
A term we often use is synergize. If you take either drug alone in a lab and expose them to malignant cells, the combination of the two drugs works better than one plus one — it’s almost one plus one plus one. You get better effects in terms of killing or limiting malignant cells. We hope to replicate in humans what we see in the lab or in mice that are engineered to have these diseases.
The data in the field has taken us towards the route of combining novel agents that have shown activity in the relapsed/refractory setting with a single agent as combination therapy upfront. Most of the data would point that if you put agent X — whether it’s selinexor, navtemadlin, pelabresib, or imetelstat — together with ruxolitinib, which tends to be the first drug you pick, you tend to see better efficacy and, in some cases, even better safety profile when the two are combined. The natural question is: if we take two active agents, can we get deeper responses? What does deeper response mean?

At the most superficial, it could mean more spleen reduction — that’s a regulatory endpoint — and deeper symptom improvements, but we’re looking at other biomarkers to understand if we’re hitting the target that we want. Are we getting deeper reductions in the driver mutation amounts of the variant allele frequency (VAF)? Are we reducing those numbers to suggest that we’re reducing the burden of disease in the bone marrow? Since we can’t measure it with a CT scan, are we reducing the disease in the bone marrow from other viewpoints like fibrosis? Is the amount of circulating abnormal cells reduced, something called the CD34+ cell number? Are we also reducing cytokines or inflammatory markers to a deeper extent?
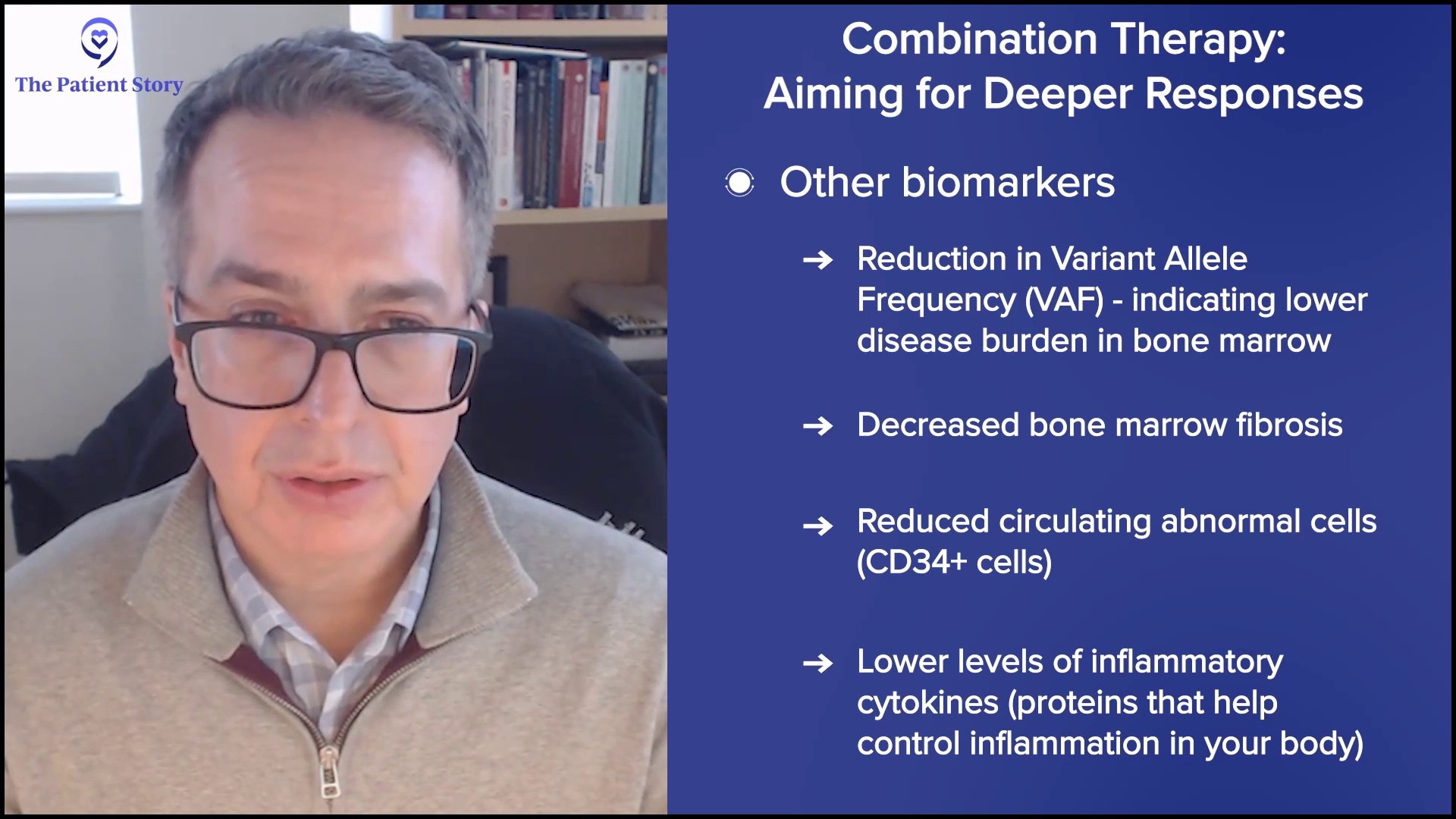
We look at all of these biological aspects and hallmarks of the disease. Are we getting even more profound effects on these biomarkers, suggesting that we’re modulating the disease more effectively? At the end of the day, MPN patients want to live better and longer, but we look for markers early in trials to understand if we’re getting there.
Andrew: These are very powerful medicines. Will the quality of life be diminished if you add this other big gun? We want to live longer, for sure, but we want to live well. Could you talk about the power of the combinations but the worry about additional side effects?

Dr. Mascarenhas: I’ll give you a prime example where one plus one can equal three from an efficacy standpoint but might still equal one from a safety standpoint. A great example is the MANIFEST-2 study. We took JAK inhibitor-naive patients with myelofibrosis and randomized them to the standard of care, which would be ruxolitinib, plus a placebo and ruxolitinib plus the study drug pelabresib, which is an oral BET inhibitor, a very rational drug that modeling has shown us should synergize very nicely with ruxolitinib. It’s a double-blinded study, so the patients and investigators don’t know what the person is getting; only a computer knows.

The answer is it did. Efficacy-wise, if you look at 24 weeks, there were very profound reductions in spleen size and very profound reductions in symptoms. If you look at the biomarkers — the bone marrow fibrosis, the inflammatory cytokines, and the JAK2 mutation — the reductions were far more significant. Everything aligned with the superiority of the combination.

But most intriguingly, if you looked at the safety profile, there were fewer grade 3 and 4 treatment-emergent side effects with the combination than with the single agent. I’ve never seen that before where two active agents combined got almost double the clinical activity and less toxicity. I hope it’s reassuring for patients that double the action doesn’t mean double the trouble. You can combine some of these drugs, get good activity, and not make patients feel worse but even make patients feel better and have less toxicity.

We have to acknowledge that we add toxicity sometimes when we add combinations. Sometimes that toxicity is in the form of gastrointestinal (GI) toxicity or lower blood counts. Sometimes it’s a trade-off. Are we getting deeper responses that could lead to better outcomes where we could be getting more cytopenia and more need for monitoring, or even transfusions? Is that reasonable for a given individual? Could we be adding some nausea and diarrhea by doing that?
What’s key to this conversation is: are we adding these toxicities continuously or periodically when some of these (MPN treatments) are dosed? For example, navtemadlin is a very active drug. When we looked at the data, it was very clear that you could get some GI toxicity. It was mostly low-grade and easy to manage, but it’s there. The drug is dosed for seven days in a row out of 28 days. The toxicity was mostly relegated to days two and nine.

This is an esoteric or personal question: for any given individual, if that deeper response could lead to better outcomes, is that period where you may have GI toxicity worth it? From a human perspective, I’m not sure. From a clinical investigation, we’re interested in trying to understand: are we providing full good at a price or is it going to be good and no price? Nothing comes for free, but these are important questions.
We rely on the patient community to tell us. We don’t simply ask patients how they’re feeling. There’s also a much-validated tool that we use called Patient Global Impression of Change (PGIC), which is simple. It asks: if you put everything together, all the toxicities that might ensue and benefits that you’re noticing or being told by the physician that you’re getting, do you feel like you are the same, a little better, a lot better, a hell of a lot better, or a little worse? The PGIC is a very valuable tool because patients will tell you if the whole thing is worth it or not. It’s key to making sure that globally, they believe that they feel that what they’re doing and what they’re going through is a net benefit at the end of the day.

Transplant vs. Medication: Where Do We Stand?
Andrew: You’ve mentioned a number of these drugs that are investigational on top of the four approved JAK inhibitors. I’m 74, so I don’t plan to do a transplant, but some patients are younger and it’s been recommended they have a transplant. As you say, it can be curative. It’s a big gun. I lived in Seattle for a long time where they developed it originally and I knew about the morbidity and mortality, and that continues for some people. Where are we now with transplant versus all these other treatments?
My hope and my goal in my lifetime and my career would be that we ultimately develop therapies that make transplants unnecessary.
Dr. John Mascarenhas
Dr. Mascarenhas: Fortunately or unfortunately, transplant remains the only modality that we have for a cure and, as you’ve pointed out, it’s not for everybody. If you’re advanced in age or have too many comorbidities, a transplant may be more dangerous and detrimental than it ever will be helpful. I’m an advocate for transplant, but it’s for a select group of patients. Patients who go into transplant are moving into an aggressive type of therapy, but it has to meet the aggressive nature of the disease. You would never take someone who has a low-risk version of the disease right into transplant because you could cause more harm earlier on than good.
It’s a complicated discussion that involves understanding where the patient is from a disease perspective, the nature of their disease, their goals, understandings, and expectations, and making sure they have a donor and support system to do a transplant. I encourage that conversation. It’s important, but it’s not going to be for everyone.

My hope and my goal in my lifetime and my career would be that we ultimately develop therapies that make transplants unnecessary. At its core, transplant is taking immune cells and using them to ultimately get rid of the grandfather or grandmother cell, which is what we call the stem cell that started the disease process, and that’s an immune-mediated elimination of the cell. If we can figure out how best to do that with medicinal therapies that may not be as intense, then we could get to a point where transplant may become historic.

At Mount Sinai, that’s what a lot of our translational research has been based on, and Ron Hoffman and others have taught me this. It’s a stem cell-directed approach to shut down or eliminate that pool of cells that allows the disease to persist. Even after you wipe out cells with a transplant, those cells can come back. Using science and collaborating with patients, targeting stem cells with rational therapies is the only way we’re going to do that.

If it weren’t for patients who show up at tertiary care centers like ours, meet physicians like me, and sign consent forms to allow us to take their blood, bank it, and use it to understand the biology, then we wouldn’t be able to move the field forward. It’s the science that we derive from our patient’s cells and their generosity, and allowing us access to their data that help us understand how to make the next generation of therapies that will target that stem cell.
Andrew: I’m a big believer in that. I go to a tertiary center as well. I’m willing to give the blood and I’d recommend that to people.

Will PV or ET Progress to Myelofibrosis? What Can Be Done?
Andrew: We have people who may not have myelofibrosis. They may have polycythemia vera or essential thrombocythemia. As they learn, they know that there can be a progression from one to another. They’re not on a JAK inhibitor, but they might be someday. What do we know about slowing progression or even knowing who will progress?
Dr. Mascarenhas: We know that the disease is chronic and progressive. Progression is not just a fear of the patient; it’s a reality that we as physicians try to risk stratify. When we meet patients, we try to understand if there are risk variables that may help us predict what that timeline might be to make treatment decisions.

We believe the rate of progression and the reason patients progress is due to clonal evolution. Blood cells acquire more mutations and alterations that allow that cell population to behave differently and change the clinical picture. We use variables, like age, anemia, white blood cell count elevation, presence of circulating blasts, low platelet count, and high molecular mutations or chromosomal abnormalities, and enter them into prognostic scoring systems, which can be found online.

Many patients will find one of these prognostic scoring systems, plug their information in, and get a sense of where they fall in prognosis. But I will caution patients: if you ever do that or speak to a physician, you must understand statistics. Please don’t make the mistake of assuming that the median survival you see is what your lifespan is. It doesn’t work that way. It’s a framework to understand where in the spectrum of patients you fall and help us make treatment decisions about the most appropriate therapy.

Our approach will evolve over time to a more refined, risk-adapted approach where we use mutations to guide us not just in the selection of therapies but the timing of when to use those therapies. From seeing enough patients, I understand that progression is a real concern for ET, PV, and myelofibrosis patients.

Progression to Acute Myeloid Leukemia (AML)
Dr. Mascarenhas: The ultimate concern about progression is the potential to evolve into acute myeloid leukemia (AML). Sometimes you might hear it called the blast phase. That’s an aggressive form of leukemia that is problematic and is a fear factor for patients and physicians treating patients with MPNs.
To see if that’s a concern, we look at mutations like p53, a type of mutation, or the presence of circulating blasts or blasts, which are immature cells in the bone marrow. That information can help us get a sense of what the risk may be of a patient transforming into an acute myeloid leukemia.
MPN-related AML is molecularly distinct from de novo AML… Unfortunately, it’s often more resistant or refractory to the traditional types of therapies that we give in that setting.
Dr. John Mascarenhas
Are We Making Progress with Secondary AML in MPN Patients?
Andrew: There’s a percentage of MF patients who traditionally would progress to AML. There’s been progress in what you’d call primary AML and there have been a number of drugs developed, but secondary AML that would come out of myelofibrosis, I understand, has been more difficult. Where are we with that now?
Dr. Mascarenhas: You’re right. For what we call de novo AML, we have a plethora of different agents. They’re still not enough, but we have agents that can be quite effective in trying to control that type of AML and induce a response. Those agents typically are not as effective and don’t have a very significant impact on the disease process in secondary AML or AML that arises out of an antecedent myeloproliferative neoplasm, whether that’s ET, PV, or myelofibrosis.
MPN-related AML is molecularly distinct from de novo AML. It looks different from a mutation profile and behaves differently. Unfortunately, it’s often more resistant or refractory to the traditional types of therapies that we give in that setting. There’s no benefit to an AML patient who had an MPN previously by giving them induction chemotherapy unless that’s followed by a transplant. Every study tells us that doesn’t improve patient outcomes, so we know that’s not effective.

We rely on drugs like hypomethylating agents, like decitabine, decitabine+cedazuridine, or azacitidine. These epigenetic therapies try to induce maturation of these immature cells, which is what leukemia is, or immature cells that don’t know grow up. We try to induce that maturation process so that they die a more natural death. It’s less intense, but it’s more effective with this type of AML.

There are a lot of other therapies under clinical investigation. Some of them are molecularly directed therapies. We’ve run trials with IDH inhibitors, oral drugs that specifically go after mutations that can be present in AML that arose out of an MPN, which can be quite effective. Understanding the molecular profile of that AML could inform treatment decisions. The reality is that’s an AML that’s problematic. Our real goal is to stop the process from evolving to AML because we know that treating that is complicated.

Role of Interferon for the Treatment of MPNs
Andrew: We’ve had interferon for a long time. Where does that fall in?
Dr. Mascarenhas: Interferon, which is a biologic that has been around a long time, is an interesting compound. There is a lot of data in the lab and in patients who’ve been treated on trials, particularly polycythemia vera and essential thrombocythemia, that this drug can be anti-cloning. You can see changes in blood count numbers with ET and PV, and reductions in mutation levels that are driving mutations like the JAK2 mutation. Groups have shown that a reduction in those levels correlates with better event-free survival (EFS). Events are clotting, bleeding, progression, and death in ET and PV. We see the value in that setting. Interferon’s increasing in its utility and use throughout the world.

Myelofibrosis is a little bit different. There’s probably some value there, particularly in treatment-naive early forms of the disease or prefibrotic forms. Interferon is under active clinical investigation in a global study, which is taking patients with lower-risk diseases that don’t have so much complexity and fibrosis in their bone marrow and have seen so many different types of therapies. Interferon works best early on in the disease course. Once the disease gets too complicated and too advanced, it may not be that effective.

New Drugs Being Studied in MPN Treatments Clinical Trials
Andrew: We mentioned the ASH conference. There are the European Hematology Association (EHA) meetings and other meetings that you attend as well. You mentioned selinexor and navtemadlin. There’s nuvisertib, elritercept, and DISC-071, an anti-hemojuvelin antibody. Help us understand this constellation of what’s going on.
Dr. Mascarenhas: You’ll notice certain themes. For the uninitiated, these names are somewhat frustrating because they don’t resonate. If one were to think about themes, they make sense.
What we’re trying to do in translating the understanding of the biology from the laboratory to the clinic is targeting signaling pathways. These pathways are inappropriately activated in malignant cells that continue to tell them to proliferate, secrete inflammation, and do things that they’re not supposed to do.

We have very intricate, complex, and overlapping signaling pathways in many malignancies, including myelofibrosis. Multiple signaling pathways are inappropriately activated or hyperactivated. For example, JAK inhibitors inhibit the JAK-STAT signaling pathway and you’ll notice those drugs end with “nib.” Those are small molecule inhibitors that inhibit enzymes in those signaling pathways.

Nuvisertib is a small molecule inhibitor which is a selective PIM-1 kinase inhibitor. It inhibits an enzyme in a signaling pathway that is very relevant to the biology of the disease. It has nice data so far that shows a reduction in symptoms, particularly spleen size and cytokines, as a single agent in patients who’ve already been on a JAK inhibitor.

Other drugs may try to turn on mechanisms that have been turned off, like navtemadlin and selinexor. These drugs are focused on different pathways but with one unifying theme, which is the p53 pathway. It’s the master regulator of cell fate. In a normal situation, your cells would get cues to commit suicide if they were infected or corrupted in some way and that is governed by the p53 pathway, an intrinsic pathway that limits the cell’s life. The problem is malignant cells have co-opted that system and turned it off. They turn it off in different ways. Navtemadlin and selinexor, through having different mechanisms, act on trying to turn that pathway on and encourage that cell to die.
You’ll see drugs that get into the cell that affect pathways, drugs that get into the cell that try to turn on pathways, and drugs that try to use the immune system in different ways to go after the abnormal cell.
Dr. John Mascarenhas
Then you might see a drug like a calreticulin antibody, which inevitably will get a name. You’ll notice early on that it’s just letters and numbers and as the development goes on, it becomes funny names that don’t make any sense and then ultimately, when it gets to the commercial space, it will be a catchy name that could be marketed. The mutant CALR antibody drug by Incyte is a perfect example of what will be a “mab,” a monoclonal antibody, which is targeted at the CALR mutant protein expressed aberrantly on the surface of the cell and is a marker for those abnormal cells. What better way to attack an abnormal cell than to have a selective marker? It’s like a handshake. It’s not even a drug; it’s a peptide. It’s a protein that binds that and by doing that, internalizes that protein complex, and that leads to the death of that cell selectively.

Approaches like that will be very fascinating. Those are immune-based approaches. They have BiTEs (bispecific T-cell engagers) that multiple companies are developing, which introduce T cells in a myelofibrosis patient to the abnormal cell by having two ends of that antibody — one that goes after the CALR, for example, and the other one that goes after CD3 on a T cell — and introduces the two cells together so that that T cell does what it should have done in the first place: recognize and create a response to that abnormal cell.
You’ll see drugs that get into the cell that affect pathways, drugs that get into the cell that try to turn on pathways, and drugs that try to use the immune system in different ways to go after the abnormal cell. These are the general themes.
Considering a Clinical Trial
Andrew: Some drugs are being studied at different levels. What would you say about considering enrolling a patient in a trial once there’s a clear picture of their case? Is there one of these that could line up with that? Take us through the thinking and discussion between a doctor and a patient about considering being in a trial.
Dr. Mascarenhas: I have to acknowledge that being on a trial is scary. I hear this all the time: guinea pig. You feel as though you may be experimented on. I always try to caution patients from that mentality because if you have a disease like myelofibrosis that is frightening to have in itself and can limit the quality of life and maybe even the time that you have, then it warrants consideration to do better than the MPN treatment standard of care. The standard of care, which is JAK inhibitors, has benefits for sure, but it’s incomplete for many patients.

The consideration for trials hinges on the patient’s goals and desires. Clinical trials may not be reasonable for every patient. Geography influences clinical trials. If you live in a rural area, the distance to a center offering a clinical trial may preclude you from participating. We know other factors may also get in the way of joining clinical trials, like language barriers, cultural barriers, and financial barriers.
I would encourage any patient who sees a specialist and goes to a tertiary care center to at least seek a consultation. This will help them understand what someone who does this full-time thinks about their disease, to clarify or classify their disease, and what clinical trial options exist.
Trials should make sense. You have to ask questions so that you understand what you’re getting involved in.
Dr. John Mascarenhas
I’ve been around long enough to know ruxolitinib as INCB18424 before it was Jakafi. The drug was used in the clinic and we prayed that it was going to make a big difference and it did. It wasn’t enough of a difference, but it made a big difference. I remember those brave patients who were the first patients to get on that study. They set the tone for the whole field and the development of other therapies because of what they put themselves into. Those patients needed the therapy, but what they did allowed us to prescribe the drug to a lot of patients. It has a profound impact way beyond that individual.

The reality is patients go on a clinical trial for themselves and that is what it should be, but you end up helping the greater good at the same time. You have to feel comfortable with joining a clinical trial. You have to read the consent form. You have to ask questions. What does it involve? What are the potential toxicities? What would be the consequences of participating in a trial?
I feel very comfortable saying that in 2025, any trial I’m aware of that’s offered to patients is an informed trial. These are not trials where we’re taking a random agent off a shelf, throwing it over there, and hoping that it works. These are drugs and agents that are vetted, have a rationale, and go through many layers of pre-testing before they go into humans. You have to trust your physician because if your physician is working with you, then the trial that’s offered to you would hopefully make sense. Trials should make sense. You have to ask questions so that you understand what you’re getting involved in.

Getting the Test Drug vs. a Placebo
Andrew: You mentioned that people ask if they’re going to be a guinea pig. They also ask if they’re going to get the “good stuff.” In other words, they want to know if they’re getting a sugar pill or a placebo. Could you talk about that concerning these trials for drugs that are investigational now?
Dr. Mascarenhas: If you’re in a phase 3 study that’s randomized, a computer randomizes you to arm A or arm B. Arm A may be the active clinical trial agent and arm B could be a placebo, but that has to be disclosed in a consent form. That’s essential. You can ask the physician, “Do I get the study drug or is it going to be randomized? Is it going to be a placebo?”
But a placebo is not always bad. For example, in the MANIFEST-2 study or what’s currently enrolling in the SENTRY study with selinexor, you get randomized as a JAK inhibitor-naive patient with myelofibrosis to either Jakafi plus a placebo, so you’re still getting active therapy, versus Jakafi plus selinexor.
It would be unethical to give nothing to someone who needs treatment.
Dr. John Mascarenhas
The idea is: can we build upon the success of the standard of care? That becomes important. It would be unethical to give nothing to someone who needs treatment. It’s not unethical and it’s practical to give someone who needs treatment the standard of care plus a sugar pill, which is blinded, versus the standard of care plus the study drug to ask which treatment regimen is better.
You can ask your physician, but you’ll notice that most trials will allow crossover and that becomes important. If you get the placebo, which only the computer knows, at some interval, most trials will allow you to cross over to the active compound. It becomes a question of: do I get the active combination upfront or is it delayed? That’s a nuance that I want to stress. If you’re participating in a phase 1 or phase 2 study, you’re going to get the drug. Those are not placebo-controlled studies. You should always be getting the drug in those settings.

What If I Have Other Chronic Conditions?
Andrew: Some of us have other conditions, like diabetes, high blood pressure, or a heart condition — for me, it’s chronic lymphocytic leukemia, which is fortunately pretty well-controlled— but we’re often excluded from trials depending on the condition. We understand the worry of the investigators. Is our data clean? Can you extrapolate from that? Can you talk about exclusion criteria and compassionate use?
Dr. Mascarenhas: The reality is there are biases in the way we do trials. We avoid patients who may be inappropriate and these are real patients who might be in need, but the trial may not be appropriate. These can be patients who have very significant kidney or heart dysfunction or a competing malignancy. Interestingly, CLL, which is more frequently seen in patients with MPNs, has different stages. Sometimes a study will allow patients who have a very indolent type of CLL to go on the study. But often, trials will be very particular, especially registration studies where they’re going for approval.

The last thing a study needs is to have confounding data where they’re unclear whether they’re making something unrelated to the myelofibrosis worse or that condition is making the assessment of the drug harder to appreciate. There is a tendency to exclude patients who have extremes of comorbidities so not all comorbidities. If you have grade 1 heart failure, that’s often allowed in a study because that’s a reality of life.
There is a very strict inclusion and exclusion criteria, what we call eligibility criteria, which determine the ability to put someone on a study. Most of those are there for safety reasons to avoid causing additional problems with a drug because it’s not intended for that reason. Another reason why they’re there is if you have a very messy, patient population with lots of diversity and heterogeneity in organ function, etc., it can be very difficult to assess whether the treatment we’re giving is safe or effective. To some extent, out of necessity, we create some homogeneity out of the heterogeneity.
Compassionate use… requires a lot of regulatory oversight, approval, and effort, which is not provided by a company or anyone.
Dr. John Mascarenhas
Andrew: How about compassionate use? If somebody would otherwise be excluded, could their doctor make a plea for a late-stage patient?
Dr. Mascarenhas: Compassionate use is often not that compassionate. It’s trying to get access to a drug under an investigational new drug (IND) and creating a protocol for one individual. What is not always appreciated is while that sounds great, many steps go into that and it requires a lot of regulatory oversight, approval, and effort, which is not provided by a company or anyone. It’s the physician and whatever team members he can assemble to try to do that.

Although compassionate use is an opportunity to get a drug, it’s often an opportunity plagued by the realities of a very cumbersome bureaucratic system that doesn’t make it very easy and timely to get to that drug. Although it’s there and in some cases helpful, it’s not always practical or realistic for people to get compassionate use and sometimes the access to those drugs is restricted by the FDA and/or the sponsor.
But compassionate use can sometimes be an opportunity to get a drug that wouldn’t otherwise be eligible and that’s key. If there’s a trial that allows the patient to get access, they often will not provide compassionate use because it would be felt unethical to allow someone to get access to a drug while all other patients would have to go through the normal routine of the clinical trial. This has to fit very strict criteria and is not always achievable.

The Future of Treatments for MPN Patients
Andrew: We have ASH in the rearview mirror. You have other conferences during the year and you’re speaking at a number of them. You’re actively involved in research and seeing patients as well. I want to get where your head’s at as far as promise in the field, how you feel about it, and what people can do about it.
Dr. Mascarenhas: We are way further ahead than we were in 2005, 2010, and 2015. I’ve watched the field grow in terms of our understanding, the amount of attention and support that’s provided to this rather small area of hematology from the federal government in terms of NIH grants — in which we indulge in getting to try to help move the field forward independent of pharmaceutical interests — but as well as pharmaceutical interest and philanthropy. They all help grease this machinery of translational research and understanding. We have cadres of smart, well-intentioned PhDs, MDs, and MD-PhDs that are helping us understand the biology and that is directly translating into the clinic to help us fine-tune our treatment.
We’re trying to understand how to get access to and develop multiple therapies that could be used in different subsets of patients or different combinations.
Dr. John Mascarenhas
You look at ASH or EHA and see endless abstracts of agents that we didn’t even know about or targets that we didn’t even understand 10 years ago. I have to believe that’s going to translate ultimately to better therapies and multiple therapies. I don’t think it’s going to be one therapy fits all. We’re trying to understand how to get access to and develop multiple therapies that could be used in different subsets of patients or different combinations and that is something that I do feel optimistic about. I would say we’re moving forward in 2025 and beyond in rational combinations and allowing science to drive the advancements.
You’ve known me for a long time. I always feel positive. I walk to work and I’m enthusiastic every day I come in because I believe in the mission. We’re making strides. I see it in the patients that I treat, but I believe it for the future.
What Patients Can Do to Alleviate Symptom Burden
Andrew: For a patient who has significant anemia, declining platelet counts, an enlarged spleen, or whatever symptom they’re experiencing, what can they do that could make a difference in the short term?
Dr. Mascarenhas: If you’re having symptom burden, particularly if it’s interfering with your quality of life and activities of daily living, you should see a physician who specializes in hematology and, ideally, someone who has expertise in this area.
The longer one delays starting therapy, the less likely that therapy will be effective and that the effects of that therapy will be durable.
Dr. John Mascarenhas
A JAK inhibitor is the front-line therapy to try to address those aspects. It doesn’t preclude one from looking at clinical trials that might even be combinations upfront of a JAK inhibitor plus an active agent to see if one can get even better, deeper, and earlier responses. But at the very least, a JAK inhibitor to try to address symptoms because you don’t get extra points for suffering through symptoms. It’s harder to rescue if one delays treatment. The data is very clear about that. The longer one delays starting therapy, the less likely that therapy will be effective and that the effects of that therapy will be durable.
Andrew: You may live at a distance from one of the major centers where there is an MPN specialist, but there are MPN specialists. You refer to cadres of researchers and physicians who are working in this area and it makes sense to have a consultation with somebody like Dr. Mascarenhas at Mount Sinai or my doctor, Dr. Jamieson at UC San Diego. Fortunately, many others are studying this and working on it and understand these nuances, so you get personalized care for where you are now and where you may be headed.

Final Takeaways on MPN Treatments
Andrew: This has been a helpful discussion. Is there anything you wanted to add, John, that we didn’t cover?
Dr. Mascarenhas: We covered all the major points and I conveyed my continued optimism. One boon that we have is it’s a very collaborative field. When you look at the abstracts, trials, and studies, you will see all the names that are listed, which reflects our collaborative nature. These are rare diseases. You can’t work in a silo. We’re all friends and colleagues. We all have a unified mission. We’re focused on that as an international team and that’s why I remain enthusiastic that we’re going to make the progress.
Andrew: Thank you for your collaboration and people. I’m seeing a lot of younger physicians and scientists interested in this area, so that gives us a great deal of hope. Dr. Mascarenhas, thank you for your devotion to us. We appreciate your time. We’re all bonded in this together. Remember: knowledge can be the best medicine of all.
Conclusion
Tiffany: That discussion [ on MPN treatments ] was the definition of news you can use. Thank you, Dr. Mascarenhas and our patient advocate and moderator Andrew, for taking the time out of your busy schedules to keep The Patient Story community informed. If you are a patient, caregiver, partner, or advocate, consider being a voice leader in your community or with us at The Patient Story.
To be empowered is to be inspired. We want you to make informed decisions about your care and that includes being educated about the latest treatment options.
MPN Patient Stories

From Confusing Symptoms to Motherhood and Advocacy: Taja’s Polycythemia Vera Story

30 Years Living with an MPN: Jeremy’s PV and Myelofibrosis Story

Directing My Life with Polycythemia Vera: A Filmmaker’s Story
